- Forum
- General C++ Programming
- Repeating Patterns
Repeating Patterns
Hello everyone, I want get opinion about constructing a function which can identify and count the "repeating n-digit sequences of numbers/characters" in a string. I guess these examples would clarify my purpose.
Suppose you have a char sequence like a DNA molecule which has only 4 options A,C,T or G.
A small part of the example would be like ...AGCTAGCTAGTATTTACGTGATGATCGATATGCATTTTACGTGATGATCGATTCGTCTACG...
Given this long string, when I enter 15, I need a function to count the number of repetitions of every 15-digit sequence that among the string.
Lets say TTTACGTGATGATCG is the sequence I want to count the repetitions in the whole string.
Which one would be a better approach for the solution:
1. There are 4 characters available,ACTG. Take input digit_number(say N), determine the (n,4) permutations of these characters with length-N.
Ex: TTTACGTGATGATCG would be a unique sequence from the permutation of 15digit set.
Ex2: TTTACGTGATGATCA(only last char different) would be considered a different case.
Then search and count for each unique sequence from the source string.
2. Parse the whole string to a memory buffer, starting from the very beginning, capture the subsequent 15digit(DNA[0-14]), search through whole string, then move on to (DNA[1-15]) and repeat the whole process.
I'm having some thoughts about the algorithm but I'm pretty sure I miss a lot of things.
I also need to consider the memory efficiency because these DNA files are large files can be up to 2GBytes. First approach seems that it'll require a lot more memory than the other.
I'd be very much grateful if you share your opinions.
Any thoughts passing through your mind would be helpful, at least I will have a different approach to my problem. Thanks
Suppose you have a char sequence like a DNA molecule which has only 4 options A,C,T or G.
A small part of the example would be like ...AGCTAGCTAGTATTTACGTGATGATCGATATGCATTTTACGTGATGATCGATTCGTCTACG...
Given this long string, when I enter 15, I need a function to count the number of repetitions of every 15-digit sequence that among the string.
Lets say TTTACGTGATGATCG is the sequence I want to count the repetitions in the whole string.
Which one would be a better approach for the solution:
1. There are 4 characters available,ACTG. Take input digit_number(say N), determine the (n,4) permutations of these characters with length-N.
Ex: TTTACGTGATGATCG would be a unique sequence from the permutation of 15digit set.
Ex2: TTTACGTGATGATCA(only last char different) would be considered a different case.
Then search and count for each unique sequence from the source string.
2. Parse the whole string to a memory buffer, starting from the very beginning, capture the subsequent 15digit(DNA[0-14]), search through whole string, then move on to (DNA[1-15]) and repeat the whole process.
I'm having some thoughts about the algorithm but I'm pretty sure I miss a lot of things.
I also need to consider the memory efficiency because these DNA files are large files can be up to 2GBytes. First approach seems that it'll require a lot more memory than the other.
I'd be very much grateful if you share your opinions.
Any thoughts passing through your mind would be helpful, at least I will have a different approach to my problem. Thanks
Last edited on
| 1. There are 4 characters available,ACTG. Take input digit_number(say N), determine the (n,4) permutations of these characters with length-N. Ex: TTTACGTGATGATCG would be a unique sequence from the permutation of 15digit set. Ex2: TTTACGTGATGATCA(only last char different) would be considered a different case. Then search and count for each unique sequence from the source string. |
Second, what does it tell you if, while iterating over every possible 15-character CTGA string, you find that the current string is equal? Every 15-character substring of a CTGA string is itself a CTGA string, and thus tautologically contained in the set of all 15-character CTGA strings.
Your second approach will work, but processing a 2 GiB string will require something in the realm of 2 quintillion string comparisons, so you'll have to think of something better.
My suggestion:
Create a trie whose leaf nodes contain indices into the DNA sequence.
Prefixes would be the first k letters of the searched for string, up so some limit (or all the way if you choose).
I expect this would be sufficiently fast if done properly. Maybe someone else can offer a better suggestion.
Create a trie whose leaf nodes contain indices into the DNA sequence.
Prefixes would be the first k letters of the searched for string, up so some limit (or all the way if you choose).
I expect this would be sufficiently fast if done properly. Maybe someone else can offer a better suggestion.
Last edited on
| First of all, the number of unique n-character strings using an m-character alphabet is not a permutation, it's m^n. |
You are right, I'm sorry english is not my main language, I had a confusion about it.
The resultant set would have 4N items.
| Second, what does it tell you if, while iterating over every possible 15-character CTGA string, you find that the current string is equal? Every 15-character substring of a CTGA string is itself a CTGA string, and thus tautologically contained in the set of all 15-character CTGA strings. |
I am having difficulty to understand what you meant by this.
| Create a trie whose leaf nodes contain indices into the DNA sequence. Prefixes would be the first k letters of the searched for string, up so some limit (or all the way if you choose). |
Do you mean assigning either ATCG to every node, so every path from root to leaf give me a unique sequence?
| Do you mean assigning either ATCG to every node, so every path from root to leaf give me a unique sequence? |
I think I'm misunderstanding you, but yes - that's what a trie is.
Each node would contain the indices into the DNA-sequence that match the node's prefix.
I'm afraid that 4^15 = 2^30 ~= one billion lookups will still be slow in practice, even if each occurs in logarithmic time - hours?
Since you won't be modifying the trie, this sort of thing can (probably) be parallelized easily.
Last edited on
There are already various applications for sequence analysis. Yes, some of that is heavy number crunching.
BLAST https://blast.ncbi.nlm.nih.gov/Blast.cgi
might not answer your exact question, but it does use a "trie" data representation of some sort.
Suffix tree, prefix tree, ...
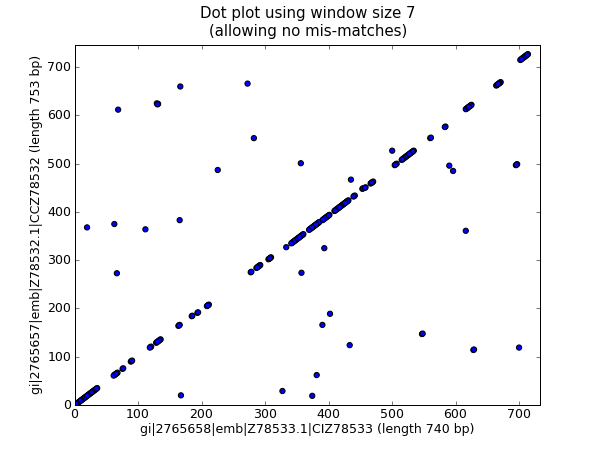
Dot plot can be used to visualize repeats. In this picture
http://biopython.org/DIST/docs/tutorial/images/dot_plot_scatter.png
there probably is a dot, if both sequences have same (N=7) pattern at the position.
(I don't know how that program, BioPython, does it, nor how it scales.)
In your case the sequence will be against itself and you need only one triagonal, without the diagonal. The dot plot creation is trivial to parallel.
Do a literature/application search to identify what algorithms already do exist for similar tasks. The best case is that an efficient function has already been written.
If you want to do "a better function", then you definitely need to know the existing methods and their pitfalls.
BLAST https://blast.ncbi.nlm.nih.gov/Blast.cgi
might not answer your exact question, but it does use a "trie" data representation of some sort.
Suffix tree, prefix tree, ...
Dot plot can be used to visualize repeats. In this picture
http://biopython.org/DIST/docs/tutorial/images/dot_plot_scatter.png
{kind=link}
there probably is a dot, if both sequences have same (N=7) pattern at the position.
(I don't know how that program, BioPython, does it, nor how it scales.)
In your case the sequence will be against itself and you need only one triagonal, without the diagonal. The dot plot creation is trivial to parallel.
| I want get opinion about constructing a function which can identify and count |
Do a literature/application search to identify what algorithms already do exist for similar tasks. The best case is that an efficient function has already been written.
If you want to do "a better function", then you definitely need to know the existing methods and their pitfalls.
Topic archived. No new replies allowed.